Current Research
Function of Autism Risk Factor Ankyrin 2 in Cortical Development and Function
Current studies focus on understanding how the high confidence autism risk gene Ankyrin2 affects cortical circuits leading to autistic behaviors such as learning and social deficits. We have a new grant from NIH to study human mutations in Ankyrin 2 to determine how they disrupt normal brain connections and function.
GABAergic Inhibitory Neurons as Targets of Cannabinoids
New studies focus on a class of GABAertic inhibitory interneuron, the basket cells, in regard to how they make synapses onto principal neurons (pyramidal neurons) in the prefrontal cortex. Synapses formed by cholecystokinin-expressing basket interneurons appear to be targets of endocannabinoids and cannabinoid drugs of abuse. How these agents affect cortical circuitry by remodeling basket cell synapses is under current investigation.
Specification of Neuronal Connectivity by Cell Recognition Molecules
How neuronal circuitry in the brain is established during development and refined in the adult is a central unanswered question in neuroscience. Neural recognition molecules expressed on the neuronal surface are pivotal players in developing cortical circuits. Among the most relevant of these molecules to human disease are members of the NCAM and L1 family (L1, CHL1, NrCAM, Neurofascin). Each of these cell recognition molecules has established functions in axon guidance that mediate correct topographic synaptic targeting of axons, while exciting new findings reveal that they also have vital functions in regulating synaptogenesis and plasticity of cortical networks.
Importantly, mutations in neural adhesion molecule genes may contribute to susceptibility to human neuropsychiatric diseases. Abnormal expression of NCAM in the developing prefrontal cortex is associated with working memory deficits in schizoprenia, while polymorphisms and aberrant splicing of NrCAM is linked to autism spectrum disorders. Mutations in the L1 gene are involved in a form of X-linked mental retardation, and CHL1 mutation is associated with low intelligence and delayed motor development. In addition to NCAM, L1 and CHL1 polymorphisms are also associated with schizophrenia in specific human populations.
To study the normal and abnormal function of neural cell adhesion molecules in brain development and function, our laboratory uses a multidisciplinary approach to generate and analyze novel mouse genetic models of neurodevelopmental disorders. We apply a panoply of methods to determine the mechanism of action of candidate genes in normal and pathological processes, including signal transduction technology, axon targeting, imaging of living neurons in acute brain slices by confocal microscopy, as well as plasmid mutagenesis and neuronal culture assays.
As a member of the Carolina Institute for Developmental Disabilities and UNC Neuroscience Center, I have productive collaborations with outstanding UNC colleagues dedicated to unraveling the neurodevelopmental mechanisms of connectivity. We are currently collaborating closely with Dr. Paul Manis, an electrophysiologist at UNC, on optogenetics and electrophysiology, and with Dr. Sheryl Moy, Director of the mouse behavioral core on working memory and fear conditioning.
Role of L1-CAMs and Semaphorins in Dendritic Spine Remodeling
In this research project we seek to illuminate a novel mechanism for dendritic spine regulation in the mammalian neocortex by immunoglobulin (Ig)-class cell adhesion molecules and secreted Semaphorins (class 3-Semas). We focused first on NrCAM (Neuron-Glial Related Cell Adhesion Molecule), a risk factor for autism spectrum disorder (ASD), and Close Homolog of L1 (CHL1), which is implicated in intellectual disability. A new concept to be studied is that L1-CAMs function as obligate subunits of Semaphorin receptor complexes to promote spine pruning in developing cortical pyramidal neurons. The central hypothesis investigated is that NrCAM, Neuropilin-2 (Npn2), and the Rap-GAP PlexinA3 (PlexA3), comprise a postsynaptic holoreceptor for Sema3F in cortical pyramidal neurons, which signals through small GTPases (Rap1, Rac1, RhoA) and PDZ scaffold and cytoskeletal effector proteins (SAP102, Ankyrin, Doublecortin-like kinase1 (DCLK1) to prune excess dendritic spines in adolescence. We will focus on the medial frontal cortex (MFC), where essential circuits regulate social behavior and cognition. The overall goal is to identify Ig-CAM-linked mechanisms that govern synaptic connectivity in the mammalian neocortex, and to elucidate how their deficiency causes abnormal brain wiring relevant to neurodevelopmental disorders.
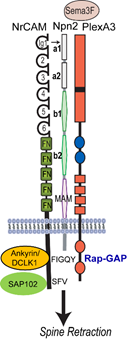
Aim 1. Developmental Regulation of NrCAM and Role of CHL1 in Dendritic Spine Remodeling
We will study a novel conditional mutant mouse (Nex1Cre-ERT2: NrCAM f/f) that inducibly deletes NrCAM from postnatal cortical pyramidal neurons to define the developmental time course for NrCAM in dendritic spine remodeling. Spines and excitatory synapses will be analyzed across early postnatal, adolescent, and adult development in pyramidal cells in the MFC of wild type (WT) and NrCAM mutants, and excitatory responses measured by whole cell recordings. Dynamics of spine loss versus gain will be studied by 2 photon live imaging in cortical slices. A novel function for CHL1 in spine remodeling will be investigated by analyzing dendritic spines, synapses, and cortical excitability in the MFC of CHL1 null mutant mice, and function in Sema3-induced spine retraction in neuronal cultures. We hypothesize that deletion of NrCAM from cortical pyramidal neurons in adolescence and loss of CHL1 will result in excess spine and synapse density, and cause cortical hyperexcitability due to impaired spine elimination.
Aim 2. Structural and Functional Analysis of NrCAM in Dendritic Spine Remodeling
Using molecular modeling and a mutagenesis approach, we will test the hypothesis that the NrCAM Ig1 domain engages the Npn2 a1 domain to form a holoreceptor complex that promotes Sema3F binding and receptor clustering/oligomerization required for spine elimination. Novel functions for NrCAM cytoplasmic domain interactions with SAP102 and cytoskeletal adaptors (Ankyrin-B, Ankyrin-G, and DCLK1) in receptor clustering and spine retraction will be probed in cortical pyramidal neurons both in vitro and in vivo. The significance of the model to be tested is that represents the first Semaphorin receptor structure that incorporates an obligate IgCAM.
Aim 3. Molecular Mechanism of Sema3F Signaling through Small GTPases
We will test the hypothesis that Sema3F-induced holoreceptor clustering stimulates PlexA3’s Rap-GAP activity, which sets in motion dual RhoA and Rac1 GTPase signaling pathways that converge at the level of the actin cytoskeleton to cause spines to collapse. Based on preliminary results, we postulate that Sema3F signaling through RhoA (Rho Kinase-Myosin II) generates contractile force that exerts tension on actin filaments assembled through Rac1 signaling (Tiam-Rac1-PAK-LIMK1-Cofilin1) to retract spines. These dual pathways will be probed using assays for GTPases, protein kinases, protein-protein interactions, and spine retraction in cortical neuron cultures. The role of intrinsic activity in Sema3F-mediated spine pruning will be evaluated by activity blockade in cortical cultures.
The outcome of these studies will have significant impact, because they will delineate a novel molecular mechanism involving IgCAMs for regulating spine and synapse development important for excitatory/inhibitory balance in frontal cortical circuits. These studies are expected to provide molecular insight into pathology associated with ASD and other neurodevelopmental disorders.
Abnormal NCAM Expression Alters GABAergic Inhibitory Neuronal Connectivity Related to Schizophrenia
A novel transgenic mouse line was generated to identify consequences on cortical development and function of expressing soluble NCAM-EC from the neuron-specific enolase promoter in developing and mature neocortex and hippocampus. NCAM-EC transgenic mice exhibited a striking reduction in synaptic puncta of inhibitory GABAergic interneurons in the cingulate, frontal association cortex, and amygdala, but not hippocampus, as shown by decreased immunolabeling of glutamic acid decarboxylase-65 (GAD65), GAD67, and the GABA transporter GAT-1. Interneuron cell density was unaltered in the transgenic mice. Affected subpopulations of interneurons included basket interneurons evident in NCAM-EC transgenic mice intercrossed with a reporter line expressing green fluorescent protein and by parvalbumin staining. Behavioral analyses demonstrated higher basal locomoter activity of NCAM-EC mice and enhanced responses to amphetamine and MK-801 compared to wild type controls. Transgenic mice were deficient in prepulse inhibition, which was restored by clozapine but not haloperidol. Additionally, NCAM-EC mice were impaired in contextual and cued fear conditioning. These results suggested that elevated shedding of NCAM perturbs synaptic connectivity of GABAergic interneurons, and produces abnormal behaviors that may be relevant to schizophrenia and other neuropsychiatric disorders.
Another project in our lab is to address a fundamental gap in understanding how an appropriate balance of excitatory and inhibitory (E/I) connectivity is achieved during development of cortical networks and adjusted through synaptic plasticity for normal functioning of the cerebral cortex. Until this gap is filled, understanding neuropsychiatric disorders with GABAergic inhibitory connection deficits, such as schizophrenia and autism, will remain a mystery. The long term goal is to identify the molecular mechanisms that establish E/I balance in the prefrontal cortex, which may identify new targets for disorders where this balance is altered. The objective is to define a novel mechanism for limiting inhibitory connections between basket interneurons and the perisomatic region of pyramidal neurons in developing prefrontal cortex. The central hypothesis is that neural cell adhesion molecule NCAM, tyrosine kinase EphA3, and ADAM10 metalloprotease comprise a presynaptic receptor complex for postsynaptic ephrinA5 that promotes elimination of perisomatic synapses critical for proper prefrontal network organization and functioning, such as in working memory.
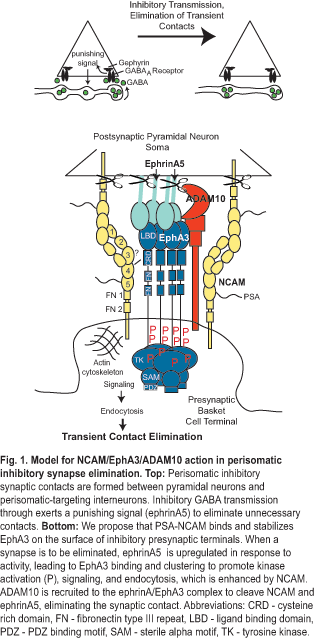
Aim 1. To identify a novel molecular mechanism for limiting perisomatic basket cell innervation in the developing mouse prefrontal cortex through NCAM-dependent ephrinA5/EphA3 signaling. We will identify NCAM/EphA3 binding sites, assess the ability of NCAM to stabilize EphA3 on the cell surface by inhibiting endocytosis and promoting ephrinA5-induced EphA3 kinase signaling, and define the developmental and activity-dependent regulation of ephrinA5 in mouse prefrontal cortex.
Aim 2. To define presynaptic and postsynaptic functions of NCAM, ephrinA5/EphA3, and ADAM10 metalloprotease in perisomatic inhibitory synapse regulation. Analysis of new conditional NCAM and ADAM10 mutant mice and cell-specific expression in brain slices will distinguish pre- versus post-synaptic functions for NCAM, ephrinA5/EphA3, and ADAM10, and test causal roles for their interactions in perisomatic synapse regulation. Dynamics of inhibitory synapse elimination will be analyzed by time-lapse two-photon microscopy in cortical slice cultures.
Aim 3. To delineate the contribution of NCAM to prefrontal cortical network organization and function using optogenetic mapping and behavioral assessment of working memory. Optogenetic mapping will be performed in brain slices from NCAM null and conditional mutant mice expressing channelrhodopsin-2 from the VGAT promoter in interneurons. Working memory performance will be measured in live mice by the delayed non-match-to-sample T-maze task.
The outcome of these studies is expected to have a sustained, positive impact, because it will illuminate novel molecular mechanisms of interneuronal connectivity that control cognitive function, while innovative optogenetic technology will elucidate cortical networks targeted in neurodevelopmental disorders.